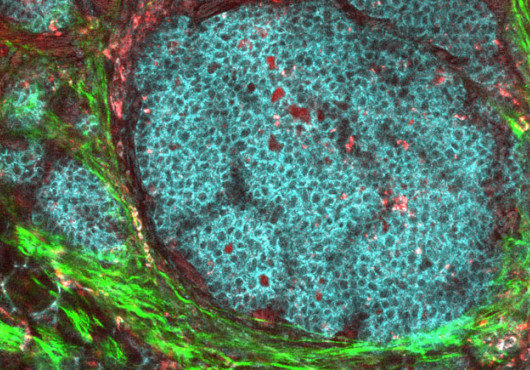
HMS researchers are one receptor closer to understanding the pathogenesis of idiopathic pulmonary fibrosis (IPF), a highly lethal disorder that scars and stiffens the lungs, impeding gas exchange.
Patients with IPF live only an average of three to five years following diagnosis and rely on lung transplants as the sole effective treatment. Many mediators have been implicated in the disorder, but the authors discovered that the bioactive lipid lysophosphatidic acid (LPA) and one of its receptors, LPA1, represent a critical pathway in the pathology. The crucial role of the LPA–LPA1 cascade was underscored by the authors’ findings that this mediator–receptor pair is largely responsible for both fibroblast recruitment and vascular leak induced by lung injury. The two processes are thought to go into overdrive when lung injury leads to fibrosis rather than repair of normal lung structures.
In the study, published in the January Nature Medicine, the researchers used bleomycin, an anti-cancer agent that can cause pulmonary fibrosis as an unwanted side effect in humans, to induce pulmonary fibrosis in wild-type and LPA1-knockout mice. Compared to wild-type mice with normal LPA1 receptors, mice lacking these receptors were protected from the deposition of collagen as well as from death produced by bleomycin lung injury, and both vascular leak and fibroblast recruitment induced by bleomycin were markedly attenuated.
Andrew Luster, senior author and the Persis, Cyrus and Marlow B. Harrison professor of medicine at HMS and Massachusetts General Hospital, and Andrew Tager, lead author and HMS assistant professor of medicine at MGH, also found elevated amounts of LPA in the bronchoalveolar lavage (BAL) fluid of patients with IPF. Further, of the five known LPA receptors, they found that only LPA1 was highly expressed on the fibroblasts found in the BAL of these patients.
The researchers reconfirmed results from earlier investigators that had shown that the BAL fluid of IPF patients contains chemoattractants that cause fibroblasts to migrate into the lung. The MGH researchers now postulated that LPA was the critical compound.
The next step featured a chemical LPA1 antagonist called Ki16425. “When we treated the responding fibroblasts with the inhibitor that targets the LPA1 receptor, it inhibited virtually all of the chemoattractant activity that was in the BAL from the IPF patients,” said Luster.
When the researchers tested the fibroblasts with another chemoattractant, the cells continued to migrate just fine. “It wasn’t that the inhibitor killed the fibroblasts or prevented them from moving to anything, it just prevented their LPA1 receptors from responding to LPA,” said Tager. The results suggest that LPA is the chemoattractant predominantly responsible for recruiting fibroblasts into the lungs of IPF patients and consequently is a new therapeutic target for the disease.