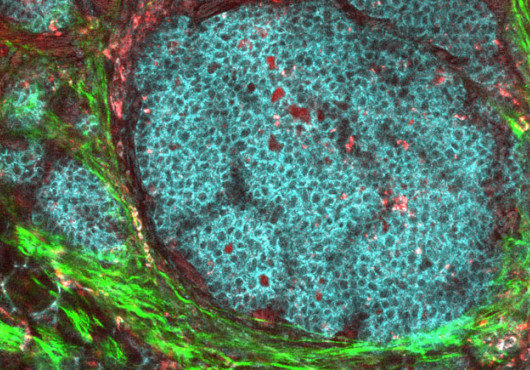
Some brain tumors develop at a leisurely pace, taking as much as 10 years before causing headaches, seizures, and other telltale symptoms. Others burst forth so quickly that a person could be healthy one month and gripped by disease the next.
“We’ve had patients come in after a car accident, and their brains appear normal on a CAT scan. They present six to eight weeks later with a massive brain lesion discovered after having a seizure,” said Ronald DePinho, HMS professor of medicine (genetics) at the Dana–Farber Cancer Institute. For years, researchers have been scrutinizing these two kinds of malignant brain tumor, or glioblastoma, to see what propels them down their distinctive hare- and tortoise-paced routes. Genetic screens have shown that the faster, or primary, glioblastomas harbor mutant versions of Pten and other genes. Defects in the well-known tumor suppressor p53 have been linked almost solely to the slower-growing, or secondary, glioblastomas.
It now appears that corrupted versions of p53 do play a role—and a common one at that—in the faster-growing variety, especially when joined by mutant versions of Pten. Hongwu Zheng, Haoqiang Ying, DePinho, and colleagues unearthed the surprising partnership using a novel tack—attempting to recreate the symptoms and pathology of primary glioblastomas in living mice. They found that they could do so by inactivating both p53 and Pten. Inactivating either p53 or Pten did not produce the brain tumors.
The researchers went back to human primary glioblastomas and found that a significant proportion harbored abnormal versions of p53 and Pten. In a further step, they discovered that the mutants conspire to raise levels of the cell-cycle protein Myc. By lowering Myc levels, Zheng and Ying, HMS research fellows in medicine, along with DePinho and colleagues, were able to stop tumor precursor cells from proliferating. The findings appear in the Oct. 23 Nature.
“You really could almost cure these cancers by bringing Myc down to physiological levels,” said DePinho.
Hopes for an effective treatment for primary glioblastoma were fanned with the recent attention on Senator Edward Kennedy’s experience with the disease, but they may be dampened by a harsh reality. Over a hundred clinical trials of new agents have been conducted in the past few years, “with virtually no impact on survival,” said DePinho. Part of the problem is that human brain cancer cells are extremely complex, often resulting from numerous mutations. Even in the mice, p53 and Pten were part of a larger network of mutations, said DePinho. Yet the discovery that the notorious pair—and mutant versions of Myc and p53 occur in a variety of tumors—may play a role in primary glioblastoma could lead to new treatments, including those that piggyback on approaches to other cancers.
When Zheng and Ying began their work, a p53–Pten partnership seemed like the oddest of couples. The researchers, who came to DePinho’s lab with the goal of identifying brain cancer–inducing partnerships, had systematically inactivated pairs of genes in mice, with little success. Though p53 was thought to be intact in primary glioblastomas, they decided to knock it out along with Pten. “I told them it was going to be a waste of time because, based on what we know, this was not a relevant mutation,” said DePinho. The effects were dramatic. “One day, the mice could be happy and jumping around. Two days later, they were totally changed,” said Zheng. Many began falling down or showing signs of seizures—classic symptoms of glioblastoma. The majority of animals, 42 out of 57, displayed symptoms, anywhere between 14 and 50 weeks after birth.
Looking at the brains of the mice, Zheng and Ying found that they harbored tumors with many of the hallmarks of human glioblastomas. Even the asymptomatic mice were found, in over half of cases, to harbor clusters of highly malignant cells closely resembling those found in humans. Intriguingly, cells in both groups, symptomatic and asymptomatic, expressed stem cell–like surface markers.
Excited by the findings, the researchers, working with colleagues at DFCI, reanalyzed 35 human primary glioblastoma samples and found that over 30 percent harbored p53 mutations. Six out of 10 also exhibited Pten mutations. Meanwhile, The Cancer Genome Atlas, a multi-institutional effort to identify genetic and epigenetic alterations in a number of different cancers, had recently identified p53 and Pten as the two most commonly mutated tumor suppressor genes in more than 200 primary glioblastomas (see Focus, Sept. 26).
Struck by the stemlike appearance of the mouse tumor cells and thinking that the p53 and Pten mutants might be working to maintain that immaturity, the researchers went back to their knockout mice. They isolated and cultured neural stem cells taken from 13-day-old embryos singly or doubly null for p53 and Pten. Compared with the single knockouts, the double mutants proliferated at much higher rates. When exposed to a differentiation-inducing medium, the single knockouts responded by turning into astrocytes, neurons, and oligodendrocytes, while the double knockouts retained their immature appearance. “They were kind of trapped in this stemlike state,” said DePinho.
A puzzle remained: how were the p53 and Pten mutants bringing about this arrest? To find out, the researchers tried once again to get the cells to differentiate,
but this time they looked at the transcripts produced by each of the knockouts. They found that 410 genes had changed in the double knockout. They then looked at the promoters of the genes and found that regions binding two transcription factors, Myc and E2F, were highly represented. Intrigued by the findings, they looked at Myc levels in the double knockout. “They were through the roof,” said DePinho.
By manipulating Myc levels in the cells, the researchers found that they could get the single and double knockouts to trade phenotypes. By lowering Myc levels, for example, they got double knockout cells to differentiate. Single knockout cells resisted differentiation when their Myc levels were raised.
In a final and compelling experiment, Zheng, Ying, DePinho, and colleagues knocked down Myc levels in human brain tumor cells and transplanted the cells into the forebrains of mice. The mice failed to develop tumors.
The results suggest that targeting Myc might be an effective strategy against brain tumors, but transcription factors are notoriously elusive targets. Gregory Verdine of Harvard University is trying to engineer peptides that can disrupt Myc’s ability to form heterodimers. “If you could find something that acts like a monkey wrench for the heterodimer, that would be a direct assault on Myc—and that would be wonderful,” said DePinho.
Students may contact Ronald DePinho at ron_depinho@dfci.harvard.edu for more information on this or other lab projects.
Conflict Disclosure: The authors report no conflicts.
Funding Sources: H. Zheng was supported by the Helen Hay Whitney Foundation. H. Ying is a recipient of the Marsha Mae Moeslein Fellowship from the American Brain Tumor Association. Grant support comes from the Goldhirsh Foundation (R.A.D.), and NIH grants U01 CA84313 (R.A.D.), RO1CA99041 (L.C.) and 5P01CA95616 (R.A.D., L.C., W.H.W., C.B. and K.L.L.). R.A.DePinho is an American Cancer Society Research Professor supported by the Robert A. and Renee E. Belfer Foundation Institute for Innovative Cancer Science.