Catching a tuberculosis infection is as easy as breathing. All it takes is inhaling a lingering droplet from a cough or sneeze of a person ill with the disease. The contagious droplet might contain only a few bacteria.
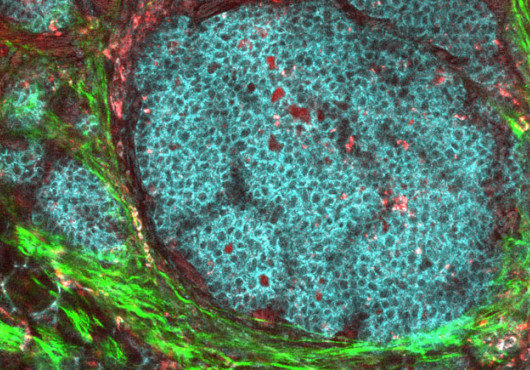
In the lung, the Mycobacterium tuberculosis (Mtb) first moves into macrophages, turning the immune cells responsible for blocking such infections into incubators. There, the bacteria slowly replicate until they burst out and spread to other macrophages.
Normally, a macrophage safely entombs bacteria in a kind of cellular shrink wrap, a new study finds. The cell dies by apoptosis, safely eliminating the pathogen. Instead, the researchers showed, virulent Mtb sabotages construction of the tough outer apoptotic envelope. The bacteria and the cell’s inflammatory contents escape through the fragile cell wall, killing the macrophage by necrosis.
“This is a mechanistic explanation for how the highly virulent tuberculosis bacterium circumvents being sequestered in an apoptotic cell,” said senior author Heinz Remold, HMS professor of medicine at Brigham and Women’s Hospital. The paper is published in the October Nature Immunology.
The paper contributes a basic insight about apoptosis that goes beyond tuberculosis, said Hardy Kornfeld, professor of medicine at University of Massachusetts Medical School, a co-author and longtime collaborator of Remold’s. “Heinz has added a new level of understanding about what might be required to make a stable apoptotic cell,” Kornfeld said. The beginning of cell death could start with hallmarks of apoptosis and turn at the last minute to necrosis, he said.
“This is a fundamentally different bacterial survival strategy than we classically consider,” said microbiologist Sarah Fortune, HSPH assistant professor of immunology and infectious diseases. “People often focus on the more proximal steps in the macrophage. This is a later question.”
Even better, these bacterial steps for intercepting host cell events can be identified and possibly countered by scientists, Fortune said. “We have thought of Mtb as the cockroach of the bacterial world. It survives in utter catastrophe. This is more evidence the bacterium is more like a puppet master, actively interfering with processes that are identifiable and targetable. This provides the groundwork for rationally designing better therapeutics.”
Ever since German physician-scientist Robert Koch first presented his discovery of the infectious agent in 1882, tuberculosis has defied efforts by scientists to prevent or cure the disease. It took more than 50 years to find the first effective treatment, the antibiotic streptomycin, to which the bacterium quickly developed resistance.
Now, the cornerstone of treatment is a drug cocktail taken daily for six to 24 months. Multidrug resistant TB infections have become a serious global health threat. The only vaccine, a disarmed strain of a bovine form of the bacterium, is largely ineffective in preventing infection, but it can prevent potentially fatal lung disease in many children.
In all but five to 10 percent of infected adults, the immune system controls, but does not eliminate, the bacteria. Chronic latent TB quietly infects an estimated one third of the world’s population. Children and people with HIV/AIDS or a compromised immune system have a higher risk of progressive disease and death. About 1.5 to 2 million people die annually from the active disease.
“We don’t know very much about the infection,” said Fortune. “We’re so far from understanding why some people get sick and some people don’t. We don’t know the molecular underpinnings that allow Mtb to survive or not or to become latent or not. We need these basic studies to identify the important processes before we can begin to understand differences between clinical groups.”
For Remold, this is a story about how discoveries in another field—apoptosis—led to new insights into the primary host cell for tuberculosis infection. “The primary infection is in the macrophage,” he said. “You have to know about the macrophage to understand the whole set of defense mechanisms against Mtb.”
Macrophages fight infections directly by engulfing and sequestering microbes and then dying by apoptosis. The same process also primes a lasting immune response, using dendritic cells as intermediaries to present antigens to the T cells.
The difference in the lungs between an effective apoptotic response to infection and necrosis is stark, said Kornfeld, a pulmonary physician who worked in the TB ward at Boston City Hospital for 20 years. “Lungs completely filled with inflammation from pneumococcal pneumonia can recover in pristine condition with treatment, because of the controlled program of apoptosis and clearance of the inflammatory response,” he said, “but even the slightest TB infection damage persists for life.”
About 10 years ago, Kornfeld, Remold, and their co-authors showed that Mtb directly induces apoptosis in human lung macrophages. Since then, they and others have shown in human-cell and mouse model studies that apoptosis seems to be good for the host and bad for Mtb. In necrosis, the reverse seems true.
This study started by testing the apoptosis phenomena observed in other systems. Remold noticed an old report on upregulated transglutaminases, enzymes that cross-link proteins to form a tough skin. Others had found a telltale grid of a lipid, phosphatidyleserine (PS), that flopped out to the cell surface in the early stages of apoptosis.
“I thought it might be interesting to find molecules that bind [to the PS template] and that could be cross-linked,” Remold said. Experiments led by first author Huixian Gan, an instructor in medicine at BWH, found the cross-linked protein annexin-1 on the surface of mouse and human apoptotic cells infected with a disarmed Mtb strain. Without annexin-1, the cells died by necrosis. Formation of the apoptotic envelope required annexin-1.
A protease inhibitor, plasminogen activator inhibitor type 2 (PAI2), protected formation of apoptotic envelope in macrophages infected with a harmless Mtb strain, but cells infected with virulent Mtb blocked PAI2 production, unleashing the action of a protease that prevented cross-linking of annexin-1 and making the unreinforced membranes vulnerable to necrosis. A final key in vivo experiment in Kornfeld’s lab, led by postdoctoral fellow Jinhee Lee, correlated lack of apoptotic envelope formation with rampant necrosis in the lungs of mice infected by the virulent Mtb strain compared with those infected by the disarmed strain.
Necrosis “fails to restrain the spread of the bacteria and also would be predicted to be ineffective in priming T cell responses that are dependent on the uptake of apoptotic vesicles by dendritic cells,” write Steven Porcelli and William Jacobs Jr. of Albert Einstein College of Medicine in the accompanying commentary. “The ability of Mtb to precisely modulate the outcome of a programmed cell death process, as shown by Gan et al., probably represents one of the important immune-evasion strategies that make this organism such a formidable challenge to global health.”
Conflict Disclosure: The authors declare no conflicts of interest.
Funding Sources: The National Institutes of Health