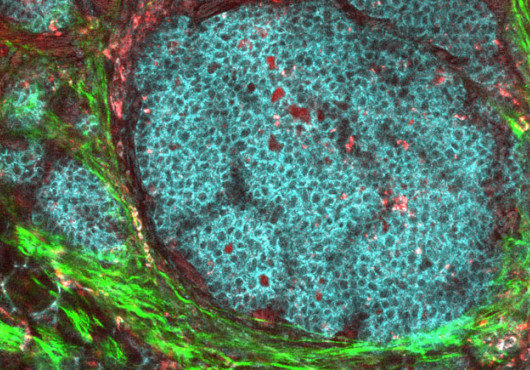
Over the last six years, the National Institutes of Health (NIH) library of publicly available genomic data has mushroomed. Researchers can now go online and peruse tens of thousands of datasets that scientists have deposited (the NIH requires that all government-funded genomic research be publicly available). But as data swells, so do the challenges: how do researchers interrogate this information torrent to find genes of interest? Forget any analogies of needles and haystacks; this is like trying to characterize every single stalk of hay from a Kansas prairie.
Now, reporting in the Aug. 6 issue of Cell Metabolism, scientists in the HMS Department of Systems Biology have developed a computational tool that can sift these massive datasets for genes of interest with remarkable speed, completing in days what normally takes one to three years. The group has also teamed up with researchers at Brigham and Women’s Hospital to verify the tool’s findings.
“We’re living in the postgenomic era now where out of 20,000 protein-coding genes only about 5,000 of them are really well studied,” said Vamsi Mootha, an HMS associate professor of systems biology and an HMS associate professor of medicine at Massachusetts General Hospital. Along with Barry Paw, HMS assistant professor of medicine at Brigham and Women’s Hospital, Mootha is co-senior author on the paper. “Now we have the opportunity to look more deeply into what the other 15,000 do,” he said.
According to Mootha and postdoctoral scientist Roland Nilsson, the vast aggregate of genomic data in the NIH depository is like a network of pools. Each pool is an individual dataset, and each dataset can contain results from hundreds of microarray experiments. Each pool, then, is swimming with countless bits of genomic clues. The trick is fishing out the particular bits you need.
To do this, Nilsson developed an algorithm that can mine this depository quickly. Starting from a handful of known genes, the algorithm hunts through all datasets and finds every gene that behaves like the initial ones.
“It’s like fishing,” said Mootha. “The initial gene set, that’s your bait. You then cast it into each pond, one at a time, and see what you catch.”
The team tested the algorithm on a series of eight genes known to be essential for hemoglobin synthesis, and fished out five genes that had never before been associated with blood production. The group then teamed up with Paw, a pediatric hematologist-oncologist, to test these findings in zebrafish. Paw and postdoctoral scientist Iman Schultz confirmed in animal models that these five genes were, in fact, essential for hemoglobin production.
“Using traditional methods, it could take well over a year to identify one of these genes,” said Paw, “whereas here, through the combined work, we can do a really quick, genomewide screen and then follow up with validation in an experimental model in a few weeks.”
Students may contact Vamsi Mootha (vamsi@hms.harvard.edu) or Barry Paw (bpaw@rics.bwh.harvard.edu) for more information.
Conflict Disclosure: The authors declare no conflicts of interest.
Funding Sources: The National Institutes of Health, the March of Dimes Foundation, the American Diabetes Association/Smith Family Foundation, and the Howard Hughes Medical Institute; the content of this work is the responsibility solely of the authors.