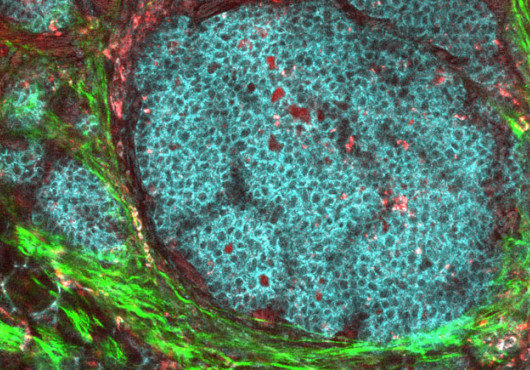
Calcium release–activated calcium (CRAC) channels are the predominant gateway for calcium entry into immune cells. Their importance is underscored by the immunodeficiency found in patients with defects in CRAC channel function.
These plasma membrane conduits open in response to calcium depletion in the endoplasmic reticulum. Two calcium sensors in the endoplasmic reticulum, STIM1 and STIM2, together with ORAI1, a CRAC channel subunit, form the major components linking endoplasmic calcium depletion to CRAC channel opening.
The laboratory of Anjana Rao, HMS professor of pathology, generated conditional knockout mice with T cell–specific disruption of Stim1, Stim2 or both, and evaluated calcium influx through CRAC channels and subsequent cytokine production.
“We didn’t know the precise mechanism of how calcium efflux from the endoplasmic reticulum store was linked to the opening of calcium channels in the plasma membrane,” explained Masatsugu Oh-hora, HMS instructor in pediatrics at the Immune Disease Institute and first author of the study, which appears in the March 9 Nature Immunology. “We were interested in understanding the function of the regulatory molecules STIM1 and STIM2. We found both proteins to be positive regulators of store-operated calcium entry in T cells.”
STIM1-deficient T cells and fibroblasts had almost no calcium influx after depletion of endoplasmic reticulum calcium stores, and the T cells failed to produce cytokines after stimulation. By evaluating whether the calcium-regulated transcription factor NFAT moved to the nucleus, the authors determined that both STIM1 and STIM2 are required to start calcium influx, while STIM2 is important for sustained calcium influx.
The authors also generated mice in which both Stim1 and Stim2 were disrupted in T cells. Analysis of the double-knockouts revealed a surprising find. The spleen and lymph nodes of the mice became massively enlarged, and immune cells were found infiltrating the lung and other organs.
“When we knocked out Stim1 and Stim2, we expected profound immunodeficiency. What we got instead was lymphoproliferative disease,” said Rao. The researchers expected that the reduction in calcium influx would affect T cell development globally; however, only one population was affected, regulatory T cells, a specialized subpopulation of T cells that act to suppress the immune system. The loss of regulatory T cells allowed other immune cell populations to expand, resulting in an aberrant immune response resembling that seen in IPEX, an X-linked human autoimmune disease that results from loss or dysfunction of a transcription factor, FOXP3. Rao’s lab previously found that FOXP3 and NFAT cooperate, explaining why defects in calcium influx that prevent NFAT from going to the nucleus might have similar effects as defects in FOXP3.
These studies advance our understanding of the link between store-operated calcium entry, T cell activation, and regulatory T cells. Further research is needed to elucidate why regulatory T cells are particularly sensitive to STIM proteins compared to conventional T cells.