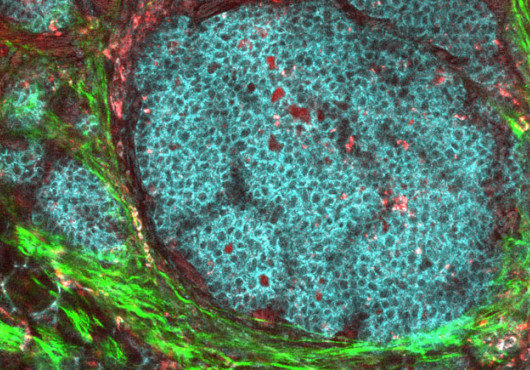
The notion that chronic inflammation causes cancer is well documented, but exactly how the disease unfolds is not always clear. New work from HSPH digs into the devilish details that link ulcerative colitis, a form of chronic inflammatory bowel disease, to colon cancer. The work hinges on a mouse model that reliably develops bowel disease and, in turn, full-blown cancer.
Because the cancer in mice closely resembles that in humans, the work opens the door to studying this complex disorder in detail. By identifying the cellular and molecular culprits that cause cancer to emerge, this work also suggests promising therapeutic approaches for colorectal cancer and ulcerative colitis and provides a robust platform for preclinical testing of possible treatments.
The work leading to this discovery, led by Laurie Glimcher, the Irene Heinz Given professor of immunology at HSPH and HMS professor of medicine at Brigham and Women’s Hospital, began in 2004 when she received reports that mice in her animal facility were sick. She investigated and found in 2007 that certain model mice developed ulcerative colitis when exposed to a specific brew of gut microflora (see Focus, focus.hms.harvard.edu/2007/101207/immunology.shtml). They dubbed the animals TRUC mice because they lack T-bet (T) and RAG2 (R), proteins known to be involved in adaptive immunity, and they develop ulcerative colitis (UC). Since then, the team has been working to identify the flora that trigger the inflammatory disease and to characterize how closely the animal disease resembles the human disease.
In addition to these efforts, Glimcher’s team acted on other evidence suggesting that their mouse model might also provide a window into studying inflammatory bowel disease–associated colon cancer. Inflammatory bowel disease is a top-three risk factor for developing colon cancer in humans. Moreover, recent research suggests that increased expression of T-bet may help increase colon cancer patient survival times. “We asked the flip question, which biologists call a reverse genetics approach,” said first author Wendy Garrett, HMS instructor in medicine at BWH. “We removed the gene of interest”—in this case the transcription factor T-bet—“and looked for the consequences.”
The repercussions were clear: studying hundreds of mice, they found that after six months, TRUC mice developed colorectal cancer. But, said Garrett, “Colorectal cancer is complicated. It’s often not just the consequence of one genetic mutation.” Rather, it is a series of genetic insults that over time becomes a vicious cycle of accumulated damage that becomes increasingly difficult to repair.
Colitis begins in the epithelium, a thin barrier just a single cell deep that separates the wastes inside the colon from the colon wall. The epithelial cells in this wall are programmed to age and die, making way for others to take their place. “Epithelial cells have a normal turnover rate,” said Garrett, which makes sense for cells so close to the toxic environment inside the colon.
In TRUC mice, immune cells in the colon wall reach through this fence, detect colitis-causing bacteria, and release inflammatory chemicals as a defense mechanism. As Glimcher and Garrett learned in 2007, this defensive strategy goes awry because the mice lack T-bet. Epithelial cells in the barrier die faster than usual, causing ulcers to form. Adding insult to injury, the presence of inflammatory cytokines, such as tumor necrosis factor alpha and reactive oxygen species, also causes DNA damage to the genes in epithelial cells.
To repair the ulcers, the body unleashes a second wave of inflammatory repair that produces more cytokines. Instead of causing cell death, these chemicals cause cell proliferation, a response intended to restore the balance between epithelial cell aging and renewal.
But in the case of chronic inflammation, this repair process backfires. The repair program causes further DNA damage and can also select for and promote the growth of very abnormal cells, tipping the balance too far in the direction of hyperproliferation of cells. “It is a perfect setup for cancer,” said Garrett, who details the work in the Sept. 8 Cancer Cell.
Because inflammation can begin when immune cells detect microbes, Glimcher and Garrett investigated whether MyD88, a molecule known to link recognition of microbes to inflammatory programs in colitis, was important in TRUC mice. They shut down MyD88, speculating that this deficit would prevent the progression of colitis. But the mice developed colitis anyway, albeit through an alternate and yet to be determined inflammatory pathway. What this shows, said Garrett, is that “there are several pathways linking microbes, inflammation and cancer.”
Since inflammation also begins because of a lack of T-bet in a subset of immune cells, Glimcher and Garrett investigated further to determine which immune cells. They found that by eliminating dendritic cells from the innate immune systems of TRUC mice, they could shut down the entire disease-causing inflammatory program. Restoring T-bet expression to the dendritic cells also shut down inflammation. In 6-month-old TRUC mice with T-bet expression restored, a few have colitis; almost none have colon cancer.
These cellular–molecular links the team discovered illuminate a clear and novel role for T-bet in dendritic cells regulating inflammation, said Glimcher, broadening the role of T-bet beyond its known role as a master regulator of the differentiation of T cells into specialized roles.
“So T-bet–based gene therapy may be something to consider,” said Glimcher. “The intestine is a good site for local delivery of therapeutics via intrarectal instillation since ulcerative colitis involves the distal part of the colon.”
Glimcher is currently looking for partners who will help her lab screen for a compound that activates T-bet. They are also working to characterize how closely the disease in mice resembles human disease. “Histologically, it looks like the human disease,” said Glimcher, “but we still don’t have all the data to know that it is, in fact, genetically and molecularly similar.” The lab is currently collecting samples of human ulcerative colitis and colorectal cancer tissue to use to make that assessment.
Students may contact Laurie Glimcher at lglimche@hsph.harvard.edu for more information.
Conflict Disclosure: Laurie Glimcher holds equity in and is on the board of directors of Bristol–Myers Squibb.
Funding Sources: The National Institutes of Health; Laurie Glimcher received support from an Ellison Scholar Award; Wendy Garrett received a Damon Runyon Cancer Research Foundation Fellowship, a Burroughs Wellcome Career in Medical Sciences Award, and funding from the V Foundation, the Dana Farber/Harvard Cancer Center, an Irving Janock Fellowship and Harvard Digestive Diseases Center Pilot Award; the authors are solely responsible for the content of this work.