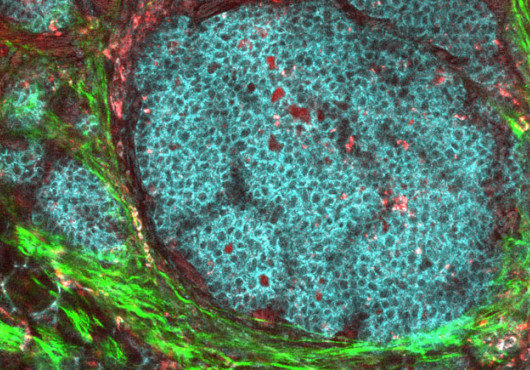
Though chronic inflammation is associated with metabolic dysfunction, the specific mechanisms underlying this connection have remained obscure. Now, a team of researchers from Chih-Hao Lee’s lab at HSPH has uncovered a molecular switch linking metabolic regulation to inflammation control. Their research appears in the June Cell Metabolism.
It was known that resident macrophages, such as those in adipose tissue, release cytokines that affect tissue function. But scientists were uncertain about how macrophage activity is modulated. To investigate, Lee’s group, led by Kihwa Kang, cocultured adipocytes or hepatocytes with macrophages to mimic their cellular crosstalk. Surprisingly, the researchers found that both hepatocytes and adipocytes release Th2 cytokines, proteins previously thought to be released only by Th2 lymphocytes.
Lee’s group further showed that Th2 cytokines target molecules within macrophages leading to changes in the macrophage phenotype. “Everyone assumed macrophages are born as M1 or M2,” Lee explained, but he suspected that some factor could be released from adipocytes to modify the immune cells.
The target receptor turned out to be peroxisome proliferator-activated receptor delta (PPAR-delta), a lipid sensor, which, when activated by Th2 cytokines, promotes the polarization necessary to change M1 macrophages into M2s. M1 macrophages are the proinflammatory, classically activated subtype that responds to bacterial infection by releasing cytokines (such as the insulin-regulating protein TNF-alpha); the M2 macrophages are anti-inflammatory and alternatively activated, responding to tissue damage as an aid in wound healing.
To test the physiological effects of disrupting the Th2 cytokine/PPAR-delta pathway, Lee’s group deleted PPAR-delta from myeloid cells in vivo. Interestingly, the PPAR-delta–lacking mice developed fatty liver and insulin-resistance, an early characteristic of type 2 diabetes and other metabolic disorders. Normally, increases in anti-inflammatory M2 macrophages keep inflammation at bay. Lee describes the pathway as a defense mechanism to balance inflammation and maintain homeostasis. “The concept is this: you have a proinflammatory signal coming out of the adipocyte once you become obese. At the same time, you have Th2 cytokine coming out as a feedback mechanism to dampen the inflammation caused by M1.”
In obese animals, adipose tissue contains more macrophages than the same tissue in lean animals, and adipocytes of obese animals release more inflammatory stimulants. Consequently, as a person grows increasingly obese, the Th2 cytokine/PPAR-delta pathway becomes overworked as it attempts to diminish inflammation. Eventually, pro-inflammatory stimulants begin to predominate as M2 macrophages and Th2 cytokines can no longer handle their regulatory functions. The balance is lost and pro-inflammatory M1 pathways take over, leading to metabolic dysfunctions including type 2 diabetes, fatty liver, and, potentially, atherosclerosis.
The group’s work does provide some therapeutic hope: by targeting the Th2 cytokine/PPAR-delta pathway that Lee and colleagues have uncovered, doctors could increase the level of M2 macrophages, which could dampen excess inflammation before it gives way to metabolic dysfunction.