When the human genome is printed out in book form, as it is in the London-based Wellcome Collection, the 4.5-point text of contiguous As, Gs, Ts, and Cs spans 118 volumes that stack neatly on 10 square meters of shelving. Though the speed at which the genome can be sequenced has increased dramatically in recent years, when it comes to reading and interpreting the sequence, we are still beginners.
Only a small part of the genome is genes. Nestled amid the rest are, among other things, binding sites for approximately 1,850 regulatory transcription factors. And only a handful of these have been characterized—through painstaking work with live cells and animal models—in terms of their locations and biological roles.
To speed this process, scientists are now turning to computational means. For the past five years, a team led by Martha Bulyk, HMS assistant professor of medicine, pathology, and health sciences and technology (HST) at Brigham and Women’s Hospital, has been devising algorithms that systematize genome parsing.
Her team recently developed PhylCRM (pronounced “fulcrum”) and Lever, a pair of algorithms that make predictions about where transcription factors bind on the genome and which genes and biological functions they regulate as a consequence. The work appears in the March Nature Methods.
“You can really apply this [algorithm] to any system,” said Bulyk, such as looking at gene sets that are up- or down-regulated during specific biological events like cell differentiation or in response to specific environmental stimuli. “It is very open-ended.”
Doing the MathAn ongoing collaboration with Alan Michelson applies these algorithms to the development of heart and muscle in the fly embryo. “We are fundamentally interested in—by whatever means is most efficient—understanding what are the transcriptional networks that regulate development,” said Michelson, senior investigator in the division of intramural research and associate director of basic research at the National Heart, Lung and Blood Institute at the NIH.
Michelson’s team applied earlier algorithms from the Bulyk lab, called ModuleFinder and Code-Finder, to a collection of genes and transcription factors expressed during different stages of fly embryo development. “If you see a transcription factor co-expressed with a set of genes in a particular developmental context—the same time and space—you infer that this transcription factor might be regulating those genes,” Michelson said.
This hypothesis, which forms the foundation of Bulyk’s algorithms, has so far been borne out. In a 2006 PLoS Computational Biology paper, Michelson and Bulyk reported that the algorithm is about 60 percent accurate in predicting enhancers belonging to one particular regulatory circuit. PhylCRM and Lever will likely improve on that by incorporating new findings.
“This is an iterative procedure,” said Michelson. “This is a way that experimentalists and computationalists can form a valuable collaboration.”
“The bottom line is that there is so much that needs to be known that cannot be immediately accessed experimentally,” said Stephen Elledge, the Gregor Mendel professor of genetics and of medicine at HMS. These algorithms can help “direct experimental science” and “instruct it.” Meanwhile, he said, experimental science can test those predictions and, in turn, help refine the algorithms.
Inside the Black BoxWhile these algorithms are continually improving, several features of the human genome make writing them thorny. Since protein-coding regions are sparse, the algorithms must search for occurrences of regulatory motifs in the remaining 95 to 99 percent of the sequence. “With that big a search space, it boils down essentially to a signal-to-noise problem,” said Bulyk. Adding to that challenge, the signal, in this case the regulatory motifs, can be as short as five base pairs. The longer the sequence and the shorter the search string, the more likely there will be spurious matches, she said.
In PhylCRM and Lever, probabilistic models and statistics, implemented by co–first authors Savina Jaeger, an HMS postdoctoral fellow, and Anthony Philippakis, a former graduate student in the lab and now a third-year MD student, help differentiate the incidental signals from the meaningful ones. These models are predominantly based on phylogenic footprinting, the concept that sequence matches across species occur because they are conserved traits.
PhylCRM scans a sequence by sliding a window across the whole genome, considering only the snippet inside the window. That snippet scores high as a candidate cis regulatory module (CRM) if it contains matches to one or more well-conserved motifs for particular transcription factors.
“Lever asks, of all the CRMs, which are most likely to be involved in turning genes on and off in specific tissues [or cell types]?” said Philippakis. Lever applies two statistical measures to determine which motif combinations and CRMs are overrepresented around genes of interest compared to background sequences, which are not thought to be involved in the biological functions of interest (see figure).
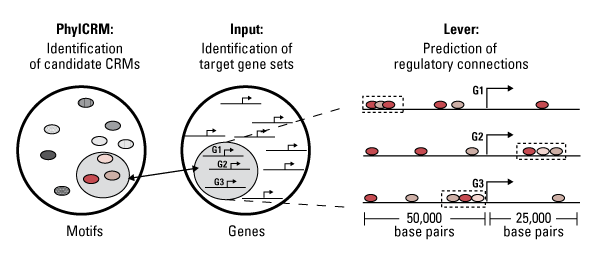
PhylCRM and Lever improve upon earlier algorithms in several ways. PhylCRM compares genomes from more species and considers their evolutionary distance. This improvement allows the algorithm to search longer sequences without losing statistical power. Lever automates the search for many motifs and motif combinations in large numbers of gene sets and the application of statistical methods to score potential regulatory relationships.
“Incorporating various types of information in these probabilistic predictive models is crucial,” said Jaeger.
Co-first author Jason Warner validated the new algorithms by applying them to muscle cell differentiation. Lever’s predictions agreed with known regulatory circuits and also identified new possibilities. Bulyk noted that Lever likely missed some regulatory connections because the algorithm favors binding sites that are conserved across genomes with perfect alignment. They are looking at ways to “allow a kind of swivel space” in future versions, she said.
While these new algorithms can be applied to many different biological scenarios, the investigators are quick to point out that there are many other parallel efforts, pursuing similar basic science. “Right now, we’re pretty bad at reading the genome. This program won’t fix that. It’s just one step along the way,” said Philippakis. The ultimate goal is the ability to translate the genome into a language scientists can understand and, perhaps, eventually use to compose their own sentences, paragraphs, chapters, and volumes.