Are some cancers benign because their cells have been lulled to sleep? Over the past few years, evidence has been building that switching on oncogenes in some cell types does not send them down the path of cancer, but rather causes them to enter the permanent sleep of senescence. In a move that is similar to the cellular suicide program of apoptosis, these cells seem to senesce as a protective mechanism, preventing them from forming a malignant cancer and causing damage to the organism.
In a study published in the December Cancer Cell, a team led by Karen Cichowski, HMS assistant professor of medicine at Brigham and Women’s Hospital, begins to unravel the mechanism underlying this phenomenon. They find that activating the oncogene Ras, a common event in cancers, causes certain cells to switch on signals that shut off the Ras pathway. This negative feedback loop seems to drive cells to inactivity.
This line of research began about a decade ago when a study found that activating oncogenes like Ras in vitro could cause cells to senesce. Cells that are senescent are inert: they no longer go through the cell cycle or respond to growth factors. Scientists debated whether this phenomenon, called oncogene-induced senescence, was real, until four papers published in Nature in 2005 showed that it occurs in vivo. “The collective hypothesis was that oncogene-induced senescence maintains a tumor in a benign state,” Cichowski said. However, it was not clear how the process works.
Rather than focusing on full-blown cancers, Cichowski’s group, led by postdoctoral fellow Stéphanie Courtois-Cox, wanted to understand the role of Ras in benign tumors. The Ras pathway is commonly mutated in cancers, but the oncogene alone is not sufficient to induce the disease. The team activated Ras in cultured cells by depleting them of a protein that normally keeps Ras in check, the NF1 tumor suppressor. Though many studies have overexpressed Ras in cells, this method has the advantage of using a natural pathway to turn on Ras at normal levels.
The team found that activating Ras in this way had surprisingly different effects in different cell types. In mouse embryonic fibroblasts, Ras activation had the expected effect of triggering several proteins known to lie downstream of it, including AKT and ERK, and these cells became immortal. But when the same experiment was performed in primary human fibroblast cells, these signals initially appeared, but then vanished. Further study showed that switching on the Ras pathway in these cells caused a rapid response from proteins that counteract the pathway. This broad negative feedback against Ras and its related proteins was soon followed by signs of senescence in the cells. “We can tell from a variety of studies that some cell types are sensitive to oncogene-induced senescence and some cell types are not,” Cichowski said. “If you have too much Ras in susceptible cells, it leads to a global negative feedback response, and this suppression helps to induce or maintain the senescent state.” One future goal is to determine what causes a cell to be sensitive or resistant to senescence signals.
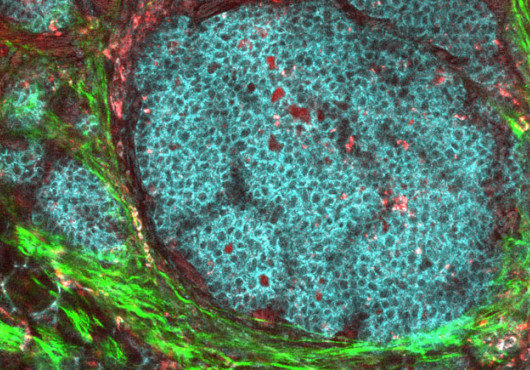
To study this phenomenon in vivo, the team examined samples from a group of patients who lack one copy of the NF1 gene and have a condition called neurofibromatosis type 1 that causes tumors to develop under the skin at peripheral nerves. Though the tumors can be painful and debilitating, most do not progress to malignant cancer. The team was able to identify senescent cells in benign tumors of NF1 patients. The senescent cells also had markers identifying them as Schwann cell progenitors, cells that are not fully differentiated and are thought to seed developing tumors in this condition. Cichowski said that it makes sense to find senescent progenitor cells, given recent evidence suggesting that cancer stem cells underlie tumor growth. “This is the first demonstration that a progenitor population was the population of cells undergoing senescence,” she said.
Kevin Shannon, the Roma and Marvin Auerback distinguished professor of pediatric molecular oncology at the University of California, San Francisco, said that this paper builds on recent research showing that “normal primary cells have a lot of defenses to protect themselves against the kinds of mutations that cause cancer.” Shannon added that the strength of a cell’s defense is weakened as the cell becomes more and more transformed into a cancer cell. When a mutation occurs in a less transformed cell, he said, the cell is better equipped to make a protective response like undergoing senescence.
Cichowski believes that cancer research has often focused on fully transformed cancer cells, rather than normal cells or benign cancers. This is studying a cell at its worst, when all the normal checks and balances that keep the cell stable are thrown off. The result is that scientists know more about cells that succumb to cancer, but little about the cells that successfully avoid it. “Studying benign tumors may give us an understanding of what needs to be deregulated” in malignant tumors, Cichowski said. Her team found that a host of proteins were active in cultured cells and in human benign tumors as part of the negative feedback against Ras, and these proteins may prove to be a guide for understanding this process and possibly developing cancer therapies.