A major challenge faced by the pharmaceutical industry has been how to rationally design and select protein molecules to create effective biologic drug therapies while reducing unintended side effects—a challenge that has largely been addressed through costly guess-and-check experiments. Researchers at the Wyss Institute for Biologically Inspired Engineering at Harvard University offer a new approach in a study published in Biophysical Journal.
“I believe that biology is the technology of this century,” said the study’s senior author Pamela Silver, the Elliott T. and Onie H. Adams Professor of Biochemistry and Systems Biology at Harvard Medical School and a Wyss Institute core faculty member. “But in order for that to be true in protein drug therapy, we must make drug discovery and development cheaper, faster and more predictable, with a higher potency for targets while eliminating side effects on healthy cells.”
Get more Harvard Medicine news here.
Merging expertise from computer science and synthetic drug design, the new model reveals that the drug efficacy of fusion-protein therapies depends on the geometric characteristics of a drug’s molecular components. Use of the model could potentially replace the need to physically make and test new biologic drug designs, reducing timelines and costs associated with drug development.
The engineered fusion proteins are created by attaching a specific antibody to a specific therapeutic protein by a “linker” made of rigid DNA strands. The antibody, a protein used as a targeting tool, is selected based on what types of cells the therapeutic portion is intended to treat. As the antibody finds its target, such as a receptor on a cancerous or otherwise infected cell, the therapeutic protein simultaneously attaches to another receptor and triggers mechanisms to disrupt the cell’s behavior. The efficacy of these new types of drugs depends on how well the two components of the fusion protein work together; that is, how well they each attach to their intended receptors at the same time.
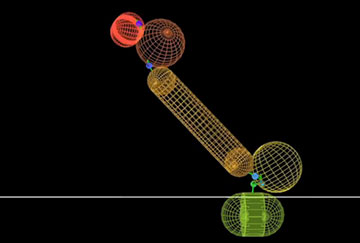
The computational model reveals that the length of the DNA linker used to connect the parts of the fusion protein influences how successfully both components are able to reach their independent receptors, empowering researchers to rationally design linker lengths that will translate into the most effective results.
“We are treating protein engineering as we would nanotechnology,” said Jeffrey Way, HMS lecturer on systems biology and senior staff scientist at the Wyss, who contributed key research to the study. “We do so by looking at biological molecules as if they are nanomachines, with programmable reactions and mechanisms.”
What’s more, the model also shows that by altering the binding strength of the therapeutic protein component, drug side effects could be further reduced. The reduced binding makes it harder for the drug to attach to cell receptors that are on the wrong cells, eliminating unintended interactions with healthy cells that share the same receptors.
“This new model helps to rationalize biologic drug discovery,” said the study’s lead author Avi Robinson-Mosher, HMS research fellow in systems biology and a postdoctoral fellow at the Wyss. “It is our vision that one day, it may be possible to perform in silico drug prospecting by using a computational model to survey a database of active proteins and targeting elements with a reasonable expectation of experimental success.” The Wyss team’s model can successfully predict how linker length, drug concentration, protein signaling strength and geometric design can impact the efficacy of a fusion protein drug, setting a new precedent in using computer simulation to potentially eliminate poor drug candidates immediately.
Moving forward, the team plans to expand the variety of drug designs the model can analyze, in hopes of creating a universal computational model that can be used as a primary investigatory tool in biologic drug development, eliminating physical assembly and experimentation as the go-to proving ground for new drug variants.
“This targeted new approach, which combines the elegance of computational design with the power of synthetic biology, offers a new way to shorten the timeline and decrease the costs involved with drug development,” said Wyss founding director Don Ingber, the Judah Folkman Professor of Vascular Biology at HMS and Boston Children’s Hospital. “But what’s more exciting is that it does this by providing a way to independently ramp up drug efficacy while tuning out toxicities, something that is not possible today.”