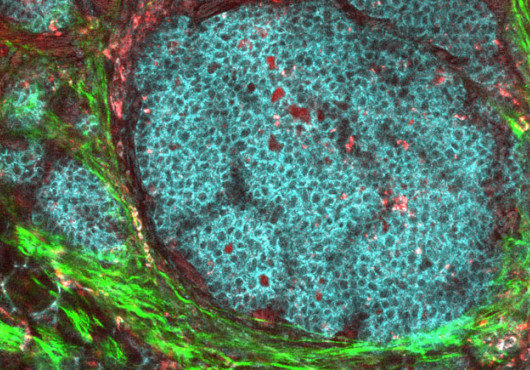
As the list of cancer-associated genetic alterations lengthens, a growing challenge is to place these alterations into signaling pathways, and the pathways into networks, in order to inform targeted drug development. Two articles in the Sept. 25 Nature describe an intersection of two major pathways in the context of colorectal cancer and, in the process, identify a potential new therapeutic target, the kinase CDK8.
Nick Dyson, HMS professor of medicine at Massachusetts General Hospital, did not set out to study colorectal cancer. He and his team were interested in the intriguing ability of the transcription factor E2F1 to promote the opposing processes of proliferation and apoptosis in normal, non-cancerous cells. Erick Morris, a postdoctoral fellow working with Dyson, noticed that when he overexpressed E2F1 in the developing Drosophila wing, specifically in newly formed epithelial cells, these cells died by apoptosis, and the result was a misshapen, gnarled wing.
To Morris and Dyson, this misshapen wing was a beautiful sight because it presented them with the opportunity to screen for regulators of E2F1-dependent apoptosis in vivo. Of the genetic interactors that emerged from this screen, one was particularly surprising: the fruit fly equivalent of beta-catenin, a key element of the Wnt signaling pathway and, significantly, a major contributor to tumorigenesis.
The observation that in Drosophila E2F1 can suppress beta-catenin suggested that beta-catenin represents a previously unsuspected point of intersection between two major signaling pathways implicated in cancer, the Wnt pathway, which stimulates beta-catenin, and the pRb pathway, which regulates E2F1.
The discovery led Morris, Dyson, and colleagues to assess E2F1 function in human cancer cells. They found that the ability of E2F1 to suppress beta-catenin activity is, indeed, conserved, and that E2F1 can directly antagonize beta-catenin–mediated transcription at target promoters such as c-myc. In addition, E2F1 activates the expression of three genes known as AXIN1, AXIN2, and SIAH1, which are known to be involved in beta-catenin degradation, further reducing the activity of the protein.
Beta-catenin is aberrantly activated in a striking 90 percent of colorectal tumors, according to Ron Firestein, an instructor in pathology working with William Hahn at Dana–Farber Cancer Institute. Recognizing the therapeutic potential of modulators of beta-catenin activity, Firestein led the search for genes whose suppression alters beta-catenin–dependent gene transcription in colon cancer cells. Using a short hairpin RNA library of more than 1,000 genes, including 95 percent of all human kinases, Firestein and colleagues identified 34 genes whose expression was necessary for beta-catenin activity.
“One of the main challenges of these high-throughput screens is identifying the true drivers of the disease among the genes that are just along for the ride,” explained Firestein. In order to sidestep this challenge, Firestein, Hahn, and colleagues conducted an additional loss-of-function screen looking for genes essential to colon cancer cell proliferation. By merging the results of these two large screens, the researchers whittled their candidates down to nine genes.
Most of the genes had not been previously associated with colorectal cancer, and the team was eager to determine whether any of them were, in fact, altered in human colorectal tumors. With the help of Adam Bass, HMS instructor in medicine at DFCI and Brigham and Women’s Hospital; Matthew Meyerson, HMS associate professor of pathology at DFCI; and colleagues, they used single nucleotide polymorphism (SNP) arrays to scan 123 human colorectal adenocarcinomas, looking for regions of DNA amplification. Among their nine genes, only one—cyclin-dependent kinase 8 (CDK8)—was located in a region that was frequently amplified in the tumors. In fact, almost 50 percent of the tumors harbored copy number gains in the CDK8 region, supporting the idea that CDK8 might be acting as an oncogene and contributing to the development of colorectal tumors.
Firestein, Hahn, and colleagues hypothesized that CDK8, a kinase involved in cell cycle progression, might affect beta-catenin transcriptional activity directly at target promoters, such as c-myc. In cultured cancer cells, they showed that CDK8 is present at the c-myc promoter, presumably as part of a multiprotein Mediator complex that acts as a bridge between sequence-specific transcription factors and the transcriptional machinery. In further support of this hypothesis, the researchers found that reducing CDK8 levels by RNA interference reduced beta-catenin binding to the c-myc promoter. The amplification of CDK8 observed in colorectal tumors, therefore, contributes to tumorigenesis at least in part by facilitating beta-catenin binding to target promoters.
Could CDK8 affect beta-catenin activity by another mechanism? Back at MGH, Dyson and his team were continuing to utilize the Drosophila model as a discovery tool. Reasoning that tumor cells might acquire a means of suppressing E2F1 as a way of maintaining higher levels of beta-catenin activity, Dyson and postdoctoral fellow Jun-Yuan Ji developed a genetic loss-of-function screen to identify additional modulators of E2F1.
Ji found that reducing E2F1 expression in the fruit fly eye perturbed the architecture of the eye, generating flies with “rough eyes.” By carefully screening for mutant flies in which this phenotype was modified, Ji identified one allele that nearly restored the eye to its normal appearance. That allele was a partially inactivated fly CDK8 gene.
“This, together with additional genetic experiments, suggested that CDK8 normally functions to antagonize E2F1 activity,” explained Ji. Further analyses by Ji and colleagues showed that CDK8 can directly interact with and phosphorylate E2F1, thereby inhibiting E2F1 activity in both Drosophila and human cancer cells.
“It’s really exciting when lines of investigation that are apparently completely independent converge on the same molecules,” said Dyson. Together, the results of these two studies indicate that CDK8 not only enhances beta-catenin transcriptional activity directly, it stimulates beta-catenin activity in a second, indirect way by releasing the protein from E2F1 repression. The upshot of both mechanisms is beta-catenin hyperactivity, leading to tumorigenesis. These findings classify CDK8 as a colorectal oncogene, and raise the question of whether CDK8 could represent a viable therapeutic target.
“Both academic and industry chemists know how to attack kinases,” said Hahn, an HMS associate professor of medicine and senior associate member of the Broad Institute, noting that inhibitors to CDK family members are already in clinical trials for other cancers.
Although the researchers caution that there is still much to be learned about the functions of CDK8 in cancer cells as well as in normal healthy cells, Hahn is optimistic about the therapeutic potential of CDK8 inhibitors. “The advantage we would have with CDK8 inhibitors in the clinic is that we could make predictions about which patients are likely to benefit,” he said, suggesting that those patients with too much CDK8 might respond best to treatment with an inhibitor.
“It’s unlikely that targeting any one molecule by itself is going to cure people,” Hahn continued, “but if you could have an armament of targeted therapies that attack two or three or four of the key pathways for a tumor, you are going to eventually get a therapy that works.”
Conflict Disclosure: William Hahn and Matthew Meyerson are consultants for Novartis (though Novartis did not support this work). Nick Dyson reports no conflict of interest.
Funding Sources: For the Dyson group: Leukemia and Lymphoma Society, Saltonstall Foundation, National Institutes of Health, MGH, Harvard Gastrointestinal Specialized Program for Research Excellence; for the Hahn group: National Cancer Institute, National Institutes of Health, Harvard–MIT Clinical Investigator Training Program, Department of Defense, Hagerty Foundation, Departament d’Educacio i Universitats de la Generalitat de Catalunya