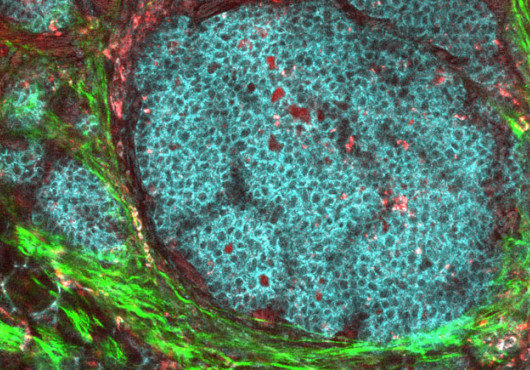
Billions of years ago, before colonies of sun-churning bacteria began filling the air with oxygen, most creatures relied on sugars to generate life-giving energy. The appearance of photosynthetic microorganisms—and later plants—would turn a huge page in Earth’s evolutionary story: the oxygen they released would become the cornerstone of a new form of energy production, one that would lead to the appearance of a dizzying array of oxygen-burning organisms. Yet the old glucose-dependent pathway would not be discarded. Though less efficient, glycolysis is fast. Even humans switch to it during periods of intense embryonic growth.
Cancer cells may also switch to the ancient glycolytic route, a phenomenon first observed nearly 80 years ago by Otto Warburg. Though it gathered dust for much of the 20th century, Warburg’s hypothesis has been attracting a lot of attention lately. Excited by the promise of a whole new pathway of potential drug targets, researchers have been working to identify the molecular mechanisms cancer cells use to make the switch. It now appears that they do so by reverting to the embryonic form of a key protein in the glycolytic pathway.
Heather Christofk, Matthew Vander Heiden, Lewis Cantley, and colleagues found that they could turn off glycolysis in cancer cell lines by replacing the embryonic (M2) version of the protein pyruvate kinase (PK) with the adult (M1) form. The substitution had an effect on the cells’ ability to develop into viable tumors: PKM1-expressing cells exhibited slower growth than those expressing PKM2. This was true not just in cell lines but in living animals. Mice receiving injections of cells expressing the PKM1 variant exhibited a delay in tumor growth compared to those receiving PKM2-containing cells. “And the tumors we got from M1 cells had all re-expressed M2,” said Vander Heiden, HMS instructor in medicine at Dana–Farber Cancer Institute. The findings appear in the March 13 Nature.
Singular DifferenceCancer cells are resolute opportunists. Rather than solve the problem of growth and survival anew, they often resurrect embryonic programs. Still, the researchers’ findings are surprising. PKM1 and PKM2 were known to differ by only 15 amino acids, but working with Ning Wu, an HMS research fellow in medicine at BID, Christofk, Vander Heiden, Cantley, and colleagues found that the critical difference may lie in a single residue. This amino acid, a lysine, sits on the lip of a deep pocket only in PMK2 (see figure). When bound by activating proteins, the lip moves aside, releasing a hidden molecule, fructose-1, 6-bisphosphate (FBP).
“It’s like popping the latch of a lid,” said Cantley, the William Bosworth Castle professor of medicine at HMS and Beth Israel Deaconess Medical Center and an HMS professor of systems biology. “There was no way we’d have predicted a single amino acid could make such a difference.”
Nor could they have foreseen the effect of this interaction. It turns out that PKM2 is turned on by FBP—the molecule is necessary for the production of energy-carrying adenosine triphosphate (ATP). So the release of FBP by the binding of activating proteins decreases energy production. “The idea that you want to shut off the glycolytic enzyme to grow is completely counterintuitive to the way I was thinking about this, and I think many people are thinking about this,” said Vander Heiden.
He, Cantley, Christofk, and their colleagues are getting used to a whole new way of thinking about the Warburg effect. Most people view the shift to the glycolytic pathway as a way to promote the synthesis of ATP. Yet, like embryonic cells, cancer cells face a daunting metabolic challenge. They must generate enormous amounts of energy to survive, grow, and proliferate, but they also must create cellular material such as DNA, RNA, proteins, carbohydrates, and lipids.
“I think a lot of people focus on ATP as the important thing, but these cells have to do both catabolism and anabolism,” said Vander Heiden. He and Cantley believe that by reverting to the nimbler embryonic form of pyruvate kinase, the cancer cell is able to switch back and forth between these two metabolic demands—the need to produce both ATP and macromolecules. Ultimately, they believe that pyruvate kinase may be under the control of growth factors, in particular, those stimulating the phosphoinositide kinase-3 (PI3K) pathway.
At Long Last, PizzazzIt was an interest in growth factors that drew the researchers to study the Warburg effect. In the late 1990s, Vander Heiden, working with Craig Thompson of the University of Pennsylvania, was studying how growth factors control a cell’s ability to survive. “It kept leading us to metabolism,” he said. Cantley, who had a long-standing interest in the PI3K pathway, was “mentally set to be interested in glycolysis.”
Working with Cantley, Christofk, then a graduate student at HMS, had been looking for binding partners for the tyrosine-phosphorylated peptides in the PI3K pathway. Said Cantley, “I asked if there was anything unusual that came out of the experiment, and she said, ‘Well, the protein that binds best to the column is pyruvate kinase.’ She said, ‘I’m not going to work on that—it’s the most boring enzyme of metabolism that I had to study as an undergraduate.’”
It would be awhile before the enzyme yielded its secrets. Christofk, currently a postdoctoral fellow at the University of California at San Francisco Cancer Center, was exploring the effect of the tyrosine-phosphorylated peptides on the M2 version of pyruvate kinase and was getting a paradoxical result: PKM2’s activity slowly decreased when the tyrosine-phosphorylated peptides were added. Wu’s crystal structure revealed the reason: the M2 pyruvate kinase was already bound to the activating molecule, FBP.
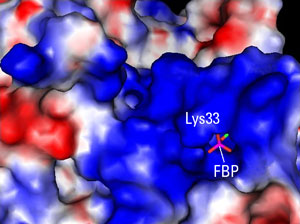
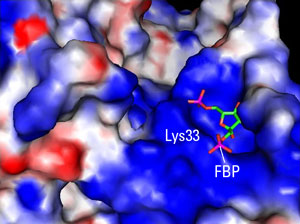
“The FBP-bound form was a big surprise because she never added any FBP during the crystallization,” said Vander Heiden. The researchers realized that the FBP was being trapped by PKM2, which was activated, and then released by tyrosine-phosphorylated proteins, thereby lowering the activity of PKM2.
“As soon as we had the structure, everything suddenly made sense,” Cantley explained.
Meanwhile, Christofk and Vander Heiden, who had joined the lab in 2005, had been creating cell lines that expressed the adult and embryonic version of pyruvate kinase and found that by knocking down PKM2, and replacing it with PKM1, they could essentially reverse the Warburg effect. Conversely, expressing PKM2 led to a restoration of glycolysis and also to faster and greater tumor growth. They injected the PKM1 and PKM2 cells into nude mice. Again, the PKM2-expressing infiltrate produced more and bigger tumors than the PKM1 cells.
On the face of it, M2 pyruvate kinase would appear ripe for targeting—and, indeed, Cantley has formed a company to explore ways to do just that. “Anything that locks PKM2 in an active or inactive mode would be a useful drug because what’s unique about PK is its ability to immediately respond to the needs of the cell,” he said.
But there is still much to learn about the already unpredictable protein. It is not clear, for example, which tyrosine phosphorylated protein binds and turns off PKM2.“There’s a lot to be discovered still,” said Cantley. “We’ve raised more questions than we’ve answered.”