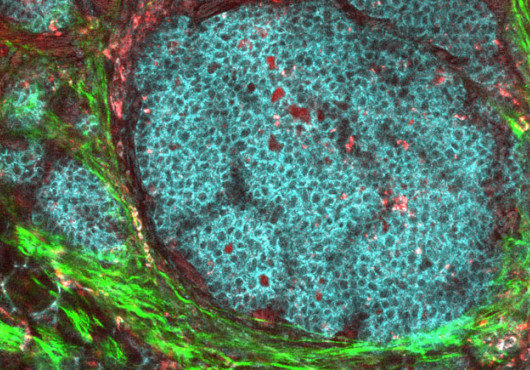
Inflammatory bowel disease (IBD) may arise from cells unable to cope with the stressful job of mediating between the army of helpful bacteria and the overprotective soldiers of the immune system.
In experiments that started in mice and extended to genetic analyses in people, a single impaired or missing gene in the intestinal tubing led to the two essential disease components. The deficit disrupted the resident bacterial community and also provoked the immune system lurking beyond the thin epithelial barrier. The findings, which appear in the Sept. 5 Cell, support a longstanding and appealing idea that epithelial cells are key players in the disease and not merely a vulnerable barrier between warring factions.
“I tell medical students that IBD results from a genetically susceptible host responding inappropriately to bacteria,” said co–senior author Richard Blumberg, gastroenterology chief at Brigham and Women’s Hospital and HMS professor of medicine. “This is evidence that the genetically mediated pathway can emanate primarily from the epithelial cells.”
IBD encompasses both ulcerative colitis (inflammation of the large intestine) and Crohn’s disease (inflammation primarily of the small intestine). Together, the painful conditions afflict about one in 2,000 people, or more than 1.5 million people in the United States.
When it is working properly, the newly implicated gene makes a transcription factor called XBP1. The gene was first discovered 20 years ago as essential to B cell differentiation in the lab of co–senior author Laurie Glimcher, the Irene Heinz Given professor of immunology at HSPH and professor of medicine at HMS. About five years ago, other groups revealed XBP1 as the mammalian equivalent of a key gene in a protective mechanism in yeast cells known as the endoplasmic reticulum (ER) stress response.
The ER stress response expands the capacity of the industrious organelle to accommodate a high protein load. The response is particularly crucial in cells that secrete a lot of proteins—such as B cells pumping out antibodies or pancreatic cells churning out digestive enzymes. Among other things, the endoplasmic reticulum translates and folds proteins and transports them to the cell surface.
In the gut, the most prolific secreting cells manage the resident bacteria by spewing antimicrobial proteins. In this study, without the Xbp1 gene, these cells became overwhelmed and died. Not only did anarchy ensue among the microbes, but the lack of Xbp1 stimulated a pro-inflammatory response in the remaining cells. “It’s a double whammy,” Glimcher said.
The study began as the latest collaboration between the Blumberg and Glimcher labs. A tantalizing paper by another group hinted that ER stress was linked to intestinal inflammation by an unknown mechanism. They teamed up to explore the connection, if any, between ER stress and the inflamed epithelial cells of IBD.
In Glimcher’s lab, postdoctoral fellow and co–first author Ann-Hwee Lee had been exploring the role of the ER stress response in mice missing Xbp1 function in the liver and pancreas. “Our hypothesis is that abnormalities in ER stress might be initiators of autoimmune disease,” Glimcher said.
In Blumberg’s lab, postdoctoral fellow and co–first author Arthur Kaser came at the study from a different angle. “Rick’s lab had a keen interest in mucosal immunology and epithelial cells,” said Kaser, an immunologist and gastroenterologist who has resumed his medical practice and research at Innsbruck Medical University in Austria.
It took only a few months to confirm their hypothesis in mice. It took a few more years for the team to work out molecular details and the relevance to human disease.
The first histology slides from mice missing the Xbp1 gene in their epithelial cells shocked Kaser. The highly secretory antimicrobial cells, called Paneth cells, were completely missing, even though the epithelial lining was intact. “We expected they would be functionally impaired, not gone,” he said. Soon, they noticed that two thirds of the mice missing both Xbp1 copies developed spontaneous inflammation of the remaining epithelial cells in small intestines in a humanlike disease. One third of the heterozygous animals suffered the same fate and were exquisitely sensitive to developing colitis.
That led to two more questions: What happened to the Paneth cells and How did inflammation develop?
XBP1 appears to be a survival factor for Paneth cells. In another conditional mouse model, researchers delayed deletion of Xbp1. Paneth cells died by apoptosis within seven days. Normally, those cells are replaced in the gut every 30 days or so. Without Paneth cells, the bacterial milieu changed. In a listeria challenge, the knockout mice had 10 times more bacteria in the gut than normal mice.
Experiments in an Xbp1-deficient mouse cell line, confirmed in vivo, showed dramatically overactivated levels of an upstream molecule called IRE1. The molecule IRE1 normally splices XBP1 to act as a transcription factor that turns on the ER stress response. As if trying to generate more XBP1, IRE1 activation increased in some kind of negative molecular feedback loop. With nothing else to do, the extra IRE1 found its other molecular dance partner, JNK, which activated a major inflammatory pathway.
But what does all this have to do with people? Three low-powered linkage studies had pointed toward some gene on chromosome 22 in the vicinity of Xbp1, Kaser found in a literature search. Up until now, this was just a valuable new mouse model of human disease. Now, the gene might have more direct biological significance for human disease. Kaser and Blumberg called in the experts: Stefan Schreiber at Christian-Albrechts-University Kiel, Germany, and his postdoctoral fellow Andre Franke.
Of all the complex diseases, human genetics is most advanced in IBD, Schreiber said. Despite this, Xbp1 does not appear among the 30 or more confirmed genes associated with Crohn’s disease in the latest round of genomewide association studies, nor is it listed in the 15 genes most strongly associated with ulcerative colitis in similarly powered studies.
A focused analysis in a cohort of 1,089 patients and 1,100 controls found a robust association with IBD, which Franke replicated in two other large cohorts. The complex haplotype suggested that rare variants may be to blame. Deep sequencing of 1,200 patients revealed more than 50 new disease variants at the Xbp1 locus.
In a mouse epithelial cell line, Lee tested two of the human variants in the coding sequence and found the same impaired molecular pathway. “The real novelty is the link of ER stress to intestinal inflammation and its relevance to human disease,” he said.
Previous genetic studies had revealed three groups of genes involved in IBD—those that deal with bacteria, those involved in the interface between innate and adaptive immunity, and those involved in nonspecific tissue destruction. “Here, suddenly, is a fourth group and a fourth principle—ER stress,” Schreiber said.
Conflict Disclosure: Laurie Glimcher is on the Board of Directors of Bristol Myers Squibb Pharmaceutical Company.
Funding Sources: Crohn’s and Colitis Foundation of America, National Institutes of Health, Ellison Medical Foundation, German National Genome Research Program (NGFN), and the DFG Excellence Cluster “Inflammation at Interfaces”